The Progression of Addison Disease: From Subclinical to Adrenal Failure
Addison Disease, a rare yet severe endocrine disorder, presents a fascinating and often challenging diagnostic journey. Far from an acute onset, this condition typically unfolds gradually, progressing from a silent, subclinical state characterized by subtle changes at the cellular level to a full-blown adrenal crisis if left undiagnosed and untreated. Understanding this progression is not just academic; it's vital for early detection, effective management, and ultimately, improving the quality of life for those affected. This article delves into the intricate stages of Addison Disease, tracing its path from the earliest molecular whispers to the overt signs of adrenal failure.
Understanding Addison Disease: A Glimpse into Adrenal Function
At the heart of Addison Disease lies the dysfunction of the adrenal glands, two small, triangular organs perched atop the kidneys. Each adrenal gland is composed of an inner medulla, responsible for producing stress hormones like epinephrine and norepinephrine, and an outer cortex. It is this outer cortex that becomes the primary battleground in Addison Disease, as it produces a suite of vital steroid hormones known as corticosteroids.
The two most critical hormones produced by the adrenal cortex, whose deficiency defines Addison Disease, are cortisol and aldosterone. Cortisol, often dubbed the "stress hormone," plays a pivotal role in regulating metabolism, suppressing inflammation, maintaining blood pressure, and managing the body's response to stress. Aldosterone, on the other hand, is crucial for maintaining electrolyte balance by regulating sodium and potassium levels, which in turn influences blood pressure and fluid volume.
Addison Disease typically manifests when at least 90 percent of the adrenal cortex has been destroyed, a testament to the adrenal glands' remarkable reserve capacity. This extensive damage leads to a significant and often debilitating reduction in the production of both cortisol and aldosterone, setting off a cascade of physiological imbalances that worsen over time.
The Roots of Adrenal Insufficiency: Key Causes and Triggers
While the progression of Addison Disease is distinct, its origins are diverse. Historically, in the mid-19th century when English physician Thomas Addison first described the condition, tuberculosis was identified as the primary culprit. Even today, tuberculosis remains a significant cause globally. However, in developed countries, the landscape has shifted dramatically.
Today, approximately 70 percent of Addison Disease cases stem from an autoimmune reaction. This occurs when the body's own immune system mistakenly targets and attacks the adrenal glands, perceiving them as a threat. Specifically, antibodies often target the 21-hydroxylase enzyme within the adrenal cortex, crucial for hormone synthesis. Autoimmune adrenal destruction can sometimes be inherited as part of a broader multiple endocrine deficiency syndrome, affecting other hormone-producing glands as well.
Beyond autoimmune attacks and tuberculosis, other infectious diseases can also lead to Addison Disease, including various fungal and viral infections. These infections often result in the calcification of the adrenal glands, impairing their function. Non-infectious causes are also significant and include adrenal hemorrhage or infarction, metastatic cancer spreading to the adrenals, congenital adrenal hyperplasia, bilateral adrenalectomy (surgical removal of both adrenal glands), and certain medications like ketoconazole, which can suppress adrenocortical function. Furthermore, conditions affecting the pituitary gland (leading to corticotropin deficiency) or the hypothalamus (causing corticotropin-releasing hormone deficiency) can indirectly result in adrenal insufficiency, though this is termed secondary or tertiary adrenal insufficiency rather than primary Addison Disease.
The Subclinical Journey: Unveiling the Early Stages of Addison Disease
The natural history of Addison Disease is characterized by a gradual decline in adrenocortical function, often referred to as the subclinical phase. This slow erosion of adrenal capacity means that individuals may experience subtle, non-specific symptoms for months or even years before a definitive diagnosis. The progression can be broadly categorized into several stages, highlighting the gradual loss of adrenal reserve and the compensatory mechanisms at play:
- Stage 0 (Potential Stage): This initial stage is characterized by the presence of adrenal autoantibodies, particularly antibodies against 21-hydroxylase, detected in the bloodstream. Crucially, at this point, all parameters of adrenal function—such as plasma ACTH, basal cortisol, ACTH-stimulated cortisol, plasma renin activity (PRA), and serum aldosterone—remain within normal limits. There are no clinical symptoms. Individuals in this stage are considered to be at high risk for developing Addison Disease in the future, making screening particularly important for those with other autoimmune conditions or a family history.
- Stage 1: As the disease subtly progresses, there's a detectable increase in plasma renin activity (PRA). While serum aldosterone might be normal or slightly low, the adrenal glands are still capable of producing an adequate cortisol response to an ACTH stimulation test. This indicates that the initial compensatory mechanisms are at play, attempting to maintain electrolyte balance despite early aldosterone insufficiency.
- Stage 2: At this juncture, the adrenal glands begin to show signs of diminished reserve. The cortisol response to an ACTH stimulation test is no longer adequate, indicating that the cortex is struggling to meet demand even when maximally stimulated. Basal cortisol levels might still be within the normal range, but the ability to mount a stress response is compromised.
- Stage 3: This stage marks a more significant decline in adrenal function. Plasma ACTH levels begin to rise significantly as the pituitary gland works harder to stimulate the failing adrenal glands (loss of negative feedback inhibition). Basal serum cortisol levels now fall to the lower limit of the normal range, and there is a virtually absent cortisol response to ACTH stimulation. Patients may begin to experience vague, non-specific symptoms such as fatigue and weakness, often mistaken for other common ailments.
Overt Adrenal Failure: Symptoms and Crisis
The final stage, Stage 4, represents overt adrenocortical failure and the manifestation of classic Addison Disease symptoms. At this point, serum or urine cortisol levels are definitively low, and plasma ACTH levels are markedly elevated. The lack of adequate cortisol and aldosterone leads to a cascade of observable symptoms that progressively increase in intensity over several months:
- Weakness and Fatigue: A profound and persistent lack of energy, often disproportionate to activity levels, is a hallmark.
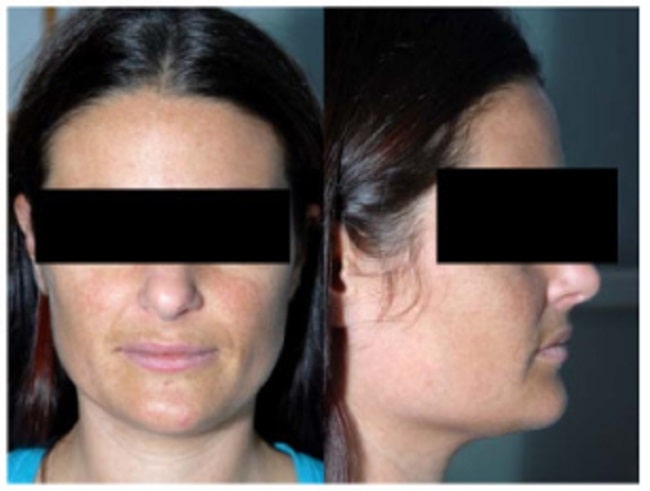
- Hyperpigmentation: A distinctive darkening of the skin and mucous membranes, particularly in sun-exposed areas, scars, skin creases, and gums, due to high ACTH stimulating melanin production.
- Gastrointestinal Upset: Nausea, vomiting, diarrhea, and abdominal pain are common.
- Poor Appetite and Weight Loss: Patients often experience a significant reduction in appetite, leading to unintended weight loss.
- Low Blood Pressure (Hypotension): Especially upon standing (orthostatic hypotension), which can cause dizziness or fainting, due to insufficient aldosterone and cortisol affecting blood volume and vascular tone.
- Salt Craving: An intense and often unusual craving for salty foods, a direct physiological response to the body's attempt to retain sodium lost due to aldosterone deficiency.
These symptoms can range from subtle to severe, eventually culminating in an adrenal crisis if left untreated. An adrenal crisis is a medical emergency characterized by sudden, severe worsening of symptoms, including profound weakness, severe pain in the abdomen, lower back, or legs, severe vomiting and diarrhea, dehydration, extremely low blood pressure, loss of consciousness, and even shock. It is a life-threatening condition requiring immediate medical intervention with intravenous corticosteroids and fluids.
Early Detection and Management: Preventing Progression
Given the insidious and progressive nature of Addison Disease, early detection is paramount. For individuals identified in the subclinical stages, particularly Stage 0 or 1 with the presence of autoantibodies, regular monitoring of adrenal function through blood tests (e.g., ACTH stimulation test, renin, aldosterone, cortisol levels) can help track progression. Identifying individuals at risk, such as those with other autoimmune conditions like Type 1 diabetes or autoimmune thyroid disease, or a family history of Addison Disease, can facilitate proactive screening.
Once diagnosed, the treatment for Addison Disease is lifelong hormone replacement therapy. This typically involves daily oral corticosteroids (e.g., hydrocortisone or prednisone) to replace cortisol and fludrocortisone to replace aldosterone. With proper medication and careful management, individuals with Addison Disease can lead full and active lives, preventing the progression to adrenal crisis and managing symptoms effectively. Education about "sick day rules" (adjusting medication during illness or stress) and carrying an emergency injection kit are also crucial aspects of self-management.
The journey of Addison Disease from its subclinical origins to overt adrenal failure is a slow but relentless one. Understanding these distinct stages, from the silent presence of autoantibodies to the dramatic presentation of an adrenal crisis, underscores the importance of vigilance and early medical intervention. While it's a chronic condition, the advancements in diagnosis and hormone replacement therapy offer a pathway to managing the disease effectively, allowing individuals to navigate its challenges with resilience and lead fulfilling lives.